
Fisiopatologia
Aunque no se ha
identificado la función precisa de la DMPK, se piensa que participa en la
reorganización del citoesqueleto, modulando la interacción entre la miosina y
la actina y de esta forma incide en la formación de las fibras de estrés y en
el tráfico de proteínas desde el retículo endoplásmico y el aparato de Golgi
hacia la membrana plasmática de las células musculares. Por lo tanto, una
deficiencia en los niveles de la DMPK podría causar alteraciones en los
procesos mencionados
Alteran, asimismo, la
expresión de los receptores de insulina (los receptores de insulina son
proteínas situadas en la superficie de las células, que les permiten responder
a la insulina), lo que explica la diabetes.

Manifestaciones clínicas
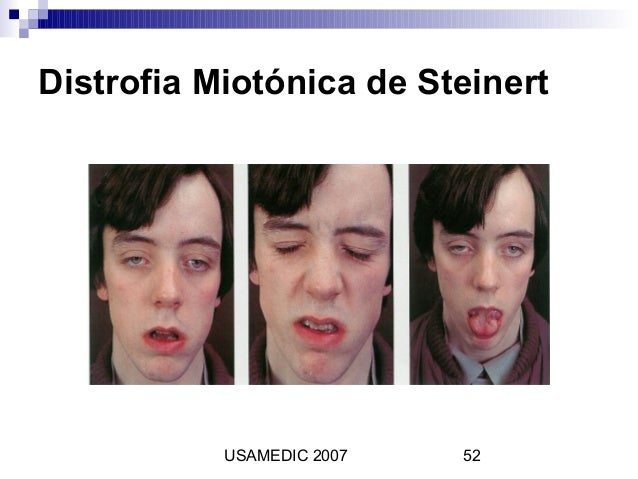
Se caracteriza por atrofia muscular predominante de los
músculos distales, axiales, faciales, faríngeos y respiratorios, miotonía en
las manos y afección multisistémica con cataratas, bloqueos de la conducción
cardiaca y arritmia, diabetes y somnolencia.
En relación con la clínica de la enfermedad hay que
considerar cuatro características importantes:
1) El carácter disociado de los signos clínicos: la
afección cardiaca puede estar aparentemente aislada, sin déficit muscular.
2) El contraste entre la severidad de la afección
cardiaca o respiratoria y la escasez o incluso ausencia de síntomas.
3) Los múltiples mecanismos causantes. En la afección
cardiaca, problemas de la conducción y/o el ritmo y más raramente disfunción
ventricular. En la afección respiratoria, afección de los músculos
respiratorios, falsas vías, hipoventilación central, embolia pulmonar.
4) Tendencia de los pacientes a una cierta apatía y a
subestimar los trastornos.
Aspectos
genéticos y herencia
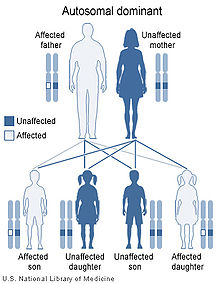
La distrofia miotónica de tipo 1 es una enfermedad
genética que se transmite de modo autosómico dominante. Una persona afectada
tiene una probabilidad del 50% de transmitir la anomalía que la origina a cada
uno de sus hijos.
La fatigabilidad muscular y los signos oculares y/o cardiacos
son indicios de este diagnóstico.
El examen ocular realizado por un oftalmólogo con la
lámpara de hendidura permite identificar las cataratas de aspecto peculiar
características de esta enfermedad.
El diagnóstico se confirma luego mediante el análisis
gené- tico (identificación de la anomalía genética asociada a la enfermedad de
Steinert), que puede realizarse a partir de una simple muestra de sangre, de
una punción de la vellosidad coriónica o del líquido amniótico.
Tratamiento
para este se realiza un tratamiento sintomatologico, solmente ademas medidas de apoyo como psicologia, psiquiatria y otros...

Bibliografia. -Distrofia Miotónica Tipo 1 o Enfermedad de Steinert / Dra. Daniela Alexandra Salvador Ramírez1, Dr. Pablo Fausto Zambrano2 intramed Vol. 3 / Número 3
No hay comentarios:
Publicar un comentario